
שיעור סיבוכים נמוך לניתוחי גירוי עצב וגאלי (Brain and Spine)
ניתוחים לגירוי עצב וגאלי מהווים התערבות יחסית בטוחה, עם שיעור סיבוכים נמוך, אם כי ישנם הבדלים בסוג הפרוצדורה ונוכחות סיבוכים,
ניתוחים לגירוי עצב וגאלי מהווים התערבות יחסית בטוחה, עם שיעור סיבוכים נמוך, אם כי ישנם הבדלים בסוג הפרוצדורה ונוכחות סיבוכים,

במאמר שפורסם בכתב העת Neurology מדווחים חוקרים על תוצאות מחקר חדש, מהן עולה כי בנשים בגיל הפוריות עם אפילפסיה כללית

מנתונים חדשים שפורסמו בכתב העת JAMA Pediatric עולה כי תוספי ויטמין C בזמן היריון עשויים להפחית את הסיכון לצפצופים בצאצאים
הפינה הפעם עוסקת בשאלה האם הטיפול ב-ADHD מזיק למטופל. למאמר המוזכר בסרטון לחצו כאן בחסות בלתי תלויה של חברת נובונורדיסק

מעבר מתרופות מניעתיות לאנגיואדמה תורשתית הניתנות בזריקה ל-Berotralstat, מעכב פומי ל-kallikrein בפלסמה, נסבל באופן כללי היטב ובעל שליטה טובה בתסמיני HAE, כך עולה ממחקר חדש שפורסם ב- Annals of Allergy, Asthma & Immunology.

כיצד ניתן לצמצם נפילות בגיל השלישי, שמהוות גורם סיכון עיקרי להדרדרות אף תמותה באוכלוסיה זו? לסקירה ב-JAMA לכל הטיפים השבועיים

בנשים עם הופעה של סוכרת הריונית לפני שבוע 20 להיריון ייתכנו תוצאות גרועות יותר, בהשוואה לנשים עם הופעה מאוחרת של

טיפול ב- Hydroxychloroquine(פלקווניל) כנגד לופוס או דלקת מפרקים שגרונית במהלך הטרימסטר הראשון להיריון אינו מביא לעליה משמעותית בסיכון למומים מולדים

נשים הנחשבות בסיכון ממוצע לחלות בסרטן שד צריכות לעבור ממוגרפיה כל שנה שנייה החל מגיל 40, במקום בגיל 50, כך עולה מההמלצות האחרונות של כוח המשימה של שירותי מניעה בארה”ב

סקר שנערך לאחרונה שתוצאותיו פורסמו בכתב העת Cancer מצא כי רק מעט רופאים עוקבים אחר המלצות החדשות של ארגון ה-ACS לבדיקות סקר לצוואר הרחם והסיבות לכך הן מגוונות

הפרעות נפשיות חמורות (serious mental illness), קשורות לסיכון מוגבר פי שניים למחלות גופניות נלוות, כך עולה ממטה-אנליזה חדשה שפורסמה ב-The Lancet Psychiatry
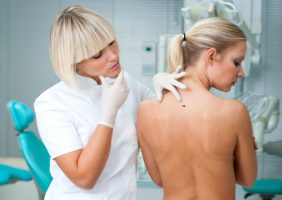
מנתונים חדשים שהוצגו במהלך הכנס השנתי מטעם ה-American Academy of Dermatology עולה קשר בין אבחנה של הידראדניטיס

במאמר שפורסם בכתב העת JAMA Network Open מדווחים חוקרים על תוצאות מחקר חדש, מהן עולה סיכון מוגבר
בקרב חולים תחת טיפול בזריקה שבועית של אגוניסטים לקולטן ל-GLP-1 שארית תכולת קיבה רבה, גורם סיכון משמעותי
מנתונים חדשים שפורסמו בכתב העת JAMA Oncology עולה כי בדיקות סקר שגרתיות המבוססות על MRI מפחיתות את

לפניכם סקירת העיתונות לחודש אפריל 2024 מאת פרופ’ יודפת, מתוך אתר החברה ליתר לחץ דם.

בגליון זה: סרטן בלוטת התריס, מאמרים אורחים, בקיצור נמרץ ועוד
גולש יקר ! ידיעה זו מיועדת לסגל הרפואי בלבד ואינה מאושרת לצפייה לקהל הרחב. אנא בצע LOG

בקיצור נמרץ – לקט מחקרים מן המחצית השניה של חודש מארס 2024

חסר בויטמין D נפוץ מאוד בילדים ובמתבגרים באופן גלובלי. בגיל הילדות חסר זה יכול להוביל לרככת, לנכות ואף לתחלואה מסכנת חיים. בעשור השני לחיים מסת העצם גדלה משמעותית ולכן גיל ההתבגרות הינו תקופה קריטית לבריאות העצם בהמשך החיים. מעבר לכך, הידע לגבי השפעותיו הגופניות של ויטמין D שאינן קשורות בבריאות העצם הולך ומתרחב. קיימת חשיבות רבה להקפדה על ערכי ויטמין D תקינים בגיל הילדות וההתבגרות. מוצגות כאן יחד עמדת המומחים באנדוקרינולוגיה מהעיתון expert opinion of endocrinology and metabolism ונייר העמדה של האיגוד לרפואת המתבגרים, שפורסם בעיתון לרפואת המתבגרים, כדי לסכם את ההמלצות הנוגעות לבירור ולטיפול בחסר בויטמין D בילדים ובמתבגרים.

בקיצור נמרץ – לקט מחקרים מן המחצית השניה של חודש מארס 2024

בגליון זה: טכיקרדיה על-חדרית, הטיפול בחסר ויטמין D בילדים, בקיצור נמרץ ועוד
במאמר שפורסם בכתב העת New England Journal of Medicine מדווחים חוקרים על תוצאות מחקר חדש בו הגיוס למחקר בשלב מוקדם בשל סוגיות הנוגעות לבטיחות ההתערבות, כאשר חיסון אימהי כנגד RSV המבוסס על Perfusion F Protein אמנם הפחית סיכון לסיבוכים משנית למחלה בדרכי נשימה תחתונות על-רקע RSV, אך לווה בעליה בסיכון ללידה מוקדמת. ד”ר ברנרד ברזילי, עורך מדור ניאונטולוגיה, משתף את המאמר ומוסיף מהערותיו
לפניכם הקלטת המפגש בהנחיית ד”ר פנינה רוטמן-פיקלני ובהשתתפות פרופ’ דויד קנדלר. לאחר הרצאתו של פרופ’ דויד קנדלר הועלו מספר מקרים

לפניכם הקלטת המפגש שהתקיים ב-26.2.24, בהנחיית ד”ר נטלי לרנר מהאיגוד הישראלי לרפואת המשפחה ובהשתתפות ד”ר נירית לב, יו”ר החברה הישראלית
לפניכם הרצאה ודיון שנערכו במסגרת קצר קולע וממוקד – סוגיות בתוספי תזונה ההרצאה בחסות חברת אלטמן

ב-1 בנובמבר התקיים וובינר שעסק בניהול טראומה ראשונית ונפשית, במסגרת מיזם “איגודים נפגשים” של האיגוד הישראלי לרפואת

בסרטון זה מדגים ומבהיר פרופ’ אלכסנדר גרינשטיין את חשיבות ואופן השימוש באפליקציית ACB לצורך הערכה ובחירה מושכלת

לפניכם הרצאות קצרות על עדכונים באימונותרפיה לטיפול בסרטן עם פרופ’ גל מרקל. בחסות בלתי תלויה של

לפניכם הרצאה במסגרת קצר קולע וממוקד – סוגיות באורולוגיה תפקודית עם ד”ר שחר אהרוני, בחסות בלתי תלויה של חברת קולופלסט
הכנס פתוח להרשמה ללא תשלום לרופאים ואנשי צוות רפואי.רופאים מישראל יוזמים כנס וירטואלי בינלאומי אשר עוסק בדרכי טיפול מניעתי (Prophylaxis)

בסרטון קצר זה מזמינה אתכם ד”ר מירב פרנקל, יו”ר משותף של הכנס “מניעה שניונית של שברים אוסטאופורוטים בישראל: כאן ועכשיו”,

קונגרס משותף בין ארה”ב לאיחוד האמירויות הערביות לתוכנית ולרישום לחצו כאן בבקשה


אם אינך רואה מייל זה תקין לחצ/י כאן 03.12.2020 – Cardio Femme Go Virtual כולנו מתמודדים עם












תגובות אחרונות